The U.S. Food and Drug Administration has begun approving for use during the COVID-19 pandemic bag resuscitators that have been converted to respiratory ventilators.
Late in March, the FDA issued an Emergency Use Authorization (EUA) that cleared the way for such modifications by eliminating a mandate that manufacturing facilities follow current good manufacturing practices (CGMP), including quality system requirements, when making ventilators under the EUA. Suspending the CGMP practices, which typically involve a significant amount of procedure documentation and record keeping, allowed non-medical manufacturers such as Ford and GM to begin ventilator production. The EUA also allowed commercially available bag resuscitators and other devices to be modified in a way that allows them to fill in as ventilators.
The first of these converted bag resuscitators have now been approved by the FDA. Among those designs getting the agency’s blessing is one developed by the University of Minnesota and produced by Boston Scientific and a second device developed and manufactured by Dan T. Moore Co. in Cleveland, Ohio.
The speed and means by which these devices were developed and approved is noteworthy. Medical devices must meet literally dozens of electrical and safety standards before their commercial introduction. The EUA for ventilators, for example, lists more than 22 IEEE and IEC standards that modified devices must adhere to. The usual means of demonstrating compliance with these technical standards is through testing by independent labs such as UL, CSA, or TÜV SÜD. But it looks as though the devices approved under the ventilator EUA won’t need to be tested by independent labs.
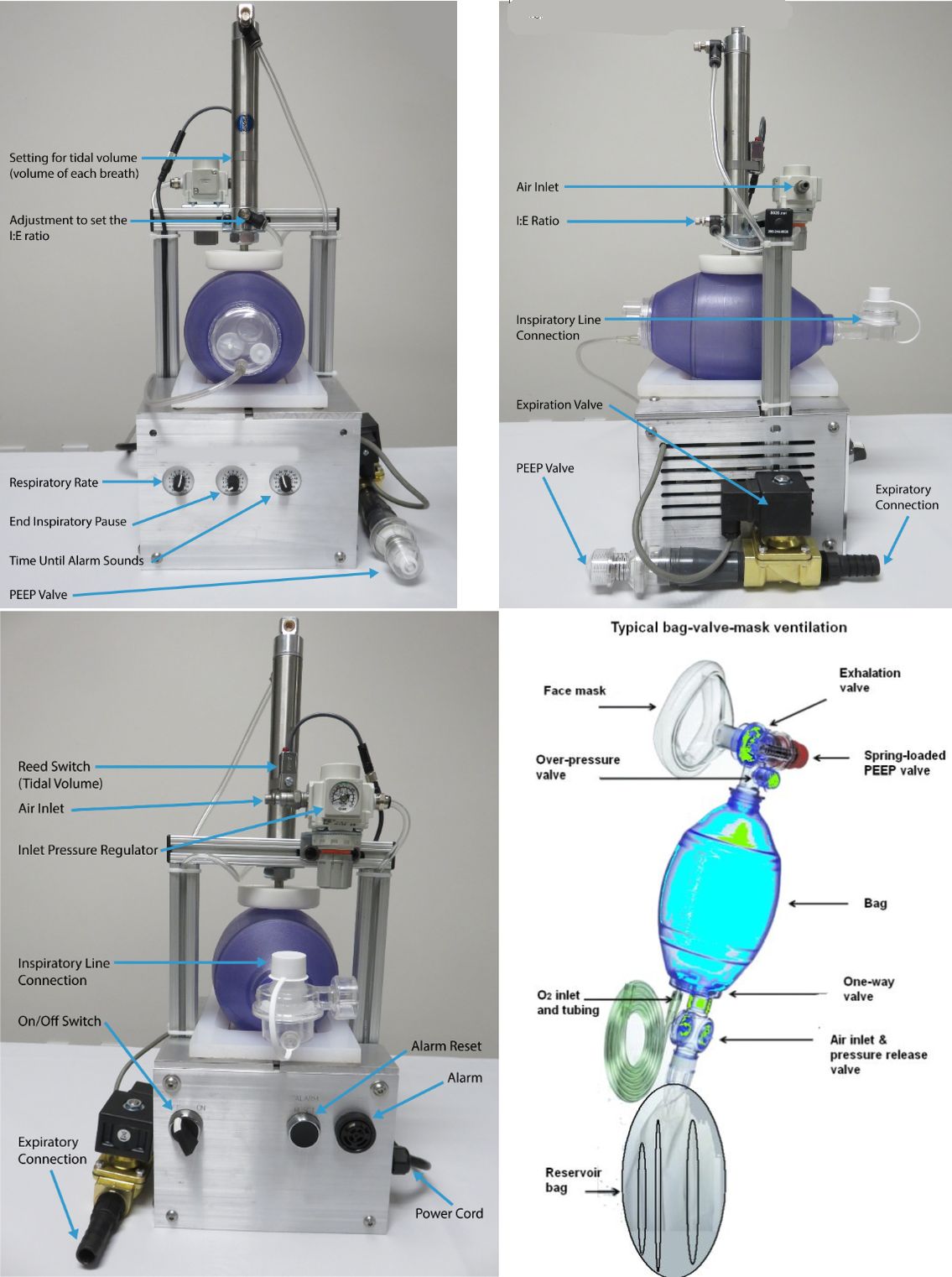
Views of the electric pneumatic resuscitator devised by SecondBreath, so named because it uses a pneumatic cylinder powered from hospital compressed air to depress the bag. It is clear from a comparison with an ordinary bag resuscitator that the process of converting these devices to respirators involves more than just adding a mechanical arm. The SecondBreath device, for example, includes alarms, controls, and regulators not found on bag resuscitators, all needed to meet ARDS Net compliance. Click image to enlarge.
Officials involved with obtaining FDA approval of the Cleveland-based effort, called SecondBreath, describe a process where device developers ran their own tests, then sent the result to the FDA for review. SecondBreath spokesman Windsor Ford says the firm’s interactions with the FDA were mainly through email. No FDA personnel inspected the manufacturing facility which consists of unused space in a Dan T. Moore warehouse. And no FDA personnel supervised testing. Nevertheless, there was “an enormous amount of testing” involved in proving the modified resuscitators complied with FDA requirements, says Ford. Some of this testing took place on a breathing simulator called an ASL 5000 which can “breath” while hooked up to a ventilator. The SecondBreath device also had to go through ARDS Net (Acute Respiratory Distress Syndrome) compliance testing which took place in cooperation with local hospitals. “We provided the FDA with extensive video and other documentation of our tests that made them comfortable that we had performed all the tests correctly,” says Ford.
One factor that simplified the FDA approval process somewhat was that almost all the components on the SecondBreath machine had UL approval for medical use. “If you have a component that doesn’t have that approval, you just have to be 100% transparent about it. The main part on our device that didn’t have that UL approval was the metal enclosure we machined ourselves,” said Ford.
Also helpful for the ventilator effort was the fact that most of the Dan T. Moore production facilities meet ISO 9000 quality management standards. “That gives us a leg up over the guy who is trying to do this in a garage,” said Ford.
It was two engineers working for Dan T. Moore Co. auto parts manufacturers who came up with the initial design. SecondBreath has already produced 30 of the devices – technically called electric pneumatic resuscitators. The company will soon be able to make between 100 and 200 daily, depending on customer demand, said Ford.
But the path to that production has been surprisingly smooth. “The FDA was amazingly helpful throughout the whole application process,” said Ford.
You may also like:


Filed Under: Medical-device manufacture, Fighting COVID-19



